

Рис. 1. Значения стационарного сечения твердой фазы

Рис. 2. Электронные микрофотографии твердых фаз, полученных в системе «вода–карбонат кальция–НТФК»с

Рис. 3. Рентгендифрактограммы твердой фазы, полученной в системе «вода–карбонат кальция–ингибитор»

Рис. 4. Результаты численного моделирования фазовых равновесий
Введение
Одной из проблем, ограничивающих практическое применение ингибиторов класса органофосфонатов (а следовательно, и эффективность эксплуатации теплотехнического оборудования), в настоящее время является образование в отдельных случаях твердых фаз, в которых наряду с солями жесткости содержатся ионы фосфонатов. С эмпирической точки зрения образование таких фаз анализируется в работах [2, 3].
В настоящей работе приведены результаты экспериментального исследования и предложена методика математического моделирования динамики конкурентного образования кристаллической и аморфной фаз из пересыщенных растворов солей щелочноземельных металлов в присутствии органофосфонатов. Считается установленным, что ингибирующее действие фосфонатов на рост кристаллов связано с их способностью адсорбироваться на поверхности кристаллов и препятствовать процессу встраивания структурных единиц кристалла в кристаллическую решетку.
В работах [4, 5] установлена критическая степень заполнения поверхности кристалла частицами ингибитора C, при которой рост кристалла прекращается. Например, в присутствии ионов нитрилотриметилфосфоната кальция N(CH2PO3)3CaH3– (НТФК) рост кристаллов карбоната кальция прекращается при C ≈ 0,1, а кристаллов сульфата бария — при C ≈ 0,16.Образование аморфных фаз в рассматриваемой области значений относительного пересыщения раствора отмечено в работах [2, 3].
При этом в работе [2]отмечено, что твердая фаза, формирующаяся в области высоких значений относительного пересыщения раствора в присутствии ингибитора, имеет приблизительно постоянный химический состав. Например, в пересыщенных растворах солей кальция в присутствии НТФК образуется аморфная фаза, химический состав которой может быть выражен эмпирической формулой N(CH2PO3)3Ca2,5H.
Растворимость данной фазы в воде при температуре 343 К и pH = 5,5 составляет 174 г/м3. Аморфная фаза является метастабильной и может переходить в кристаллическую фазу того же химического состава, однако скорость такого превращения мала, и в дальнейшем ею будем пренебрегать. Ионное равновесие между твердой аморфной фазой и водным раствором, содержащим ионы кальция, водорода и ингибитора, выраженное формальным уравнением:
2N(CH2PO3)3Ca2,5H 3Ca2+ + 2N(CH2PO3)3CaH3–,
удовлетворяет следующему условию:
(CCa,L)1,5Cinh,L = P2, (1)
где CCa и Cinh — молярные концентрации ионов, соответственно, кальция и ингибитора (НТФК), P2 ≈ const (в ограниченной области параметров системы вблизи состояния равновесия). Авторы работы [3] объясняют природу образующейся твердой фазы химическим взаимодействием ионов щелочноземельных металлов с ионами ингибитора.
В работе [7] предпринята попытка связать процессы фазообразования в системе «вода–карбонат кальция–ингибитор» с мольным соотношением Nm = Cinh/CCa . При этом авторы объясняют максимально интенсивное образование твердой фазы в определенном интервале значений Nm влиянием фиктивной турбулентности. Однако попытка связать фазообразование лишь с влиянием величины Nm = Cinh/CCa не проясняет истинной причины возникновения аморфной фазы, возникающей и растущей конкурентно с кристаллической фазой кальцита.
Привлечение для объяснения процесса формирования аморфной фазы влияния гидродинамических факторов (турбулентности) также представляется весьма произвольным, тем более, что в наших экспериментах образование аморфной фазы наблюдалось в статических условиях. Таким образом, необходимо признать, что удовлетворительное объяснение немонотонного характера зависимости количества осадка от количества вводимого ингибитора в настоящее время отсутствует.
Экспериментальная часть
Исследование распределения ионов кальция между жидкой и твердой фазами проводили путем термической обработки пересыщенного раствора гидрокарбоната кальция, имитирующего типичную весьма жесткую воду. Для приготовления пересыщенного раствора смешивали равные объемы водных растворов, один из которых содержал ионы кальция, а другой — гидро-карбонатионы.
Перед смешиванием растворов в них вводили в различных количествах ингибитор солеотложений «ИОМС1», основным компонентом которого является нейтральная натриевая соль НТФ (дополнительными примесями являются соли метил-амино-диметил-фосфоновой кислоты и соединения ряда аминов), которая может быть описана приближенной формулой N(CH2PO3)3Na3H3 и которая в водной среде взаимодействует с ионами кальция по схеме:
N(CH2PO3)3Na3H3 + Ca2+ 3Na+ 2H+ N(CH2PO3)3CaH3–,
Приготовленные таким образом образцы, имитирующие весьма жесткую воду, содержали основные компоненты в следующих концентрациях:
CCa = 27,5 моль/м3, CНСO3 =16 моль/м3,Cinh [0; 4] моль/м3,
что соответствует значениям мольного соотношения в интервале:
Nm [0; 0,15].
Эти образцы подвергали нагреванию до температуры 348 К в течение четырех часов с распадом гидрокарбонатионов:
HCO3– H+ + CO32–,
и последующим взаимодействием карбонатионов Ca32– с ионами кальция:
CO32– + Ca2+ CaCO3.
Относительное пересыщение полученного раствора карбоната кальция может быть оценено по формуле:
(T) ≈ CCaCCO3/PCaCO3(T) ~ 105.
При данном значении относительного пересыщения твердая фаза образуется во всем исследованном в настоящей работе интервале значений Cinh и, соответственно, Nm. После установления фазового равновесия проводили химический анализ, при помощи которого определяли остаточную концентрацию ионов кальция в жидкой фазе CCa,L и вычисляли коэффициент распределения кальция Ca = CCa,L/CCa.
Сечение твердой фазы (относительная доля твердой фазы в общем объеме системы) вычисляли по формуле: S = (1 – Ca)CCa/CCa,S,где CCa,S = 29 000 моль/м3 — молярная концентрация кальция в твердой фазе, определенная по результатам рентгеноструктурного анализа. Полученная в результате серии экспериментов зависимость S = f(Nm) графически представлена на рис. 1 (синие точки).
Таким образом, немонотонный характер исследуемой зависимости объективно подтверждается экспериментом и требует соответствующего теоретического объяснения. Структуру образующихся твердых фаз исследовали методами электронной микроскопии и рентгендифрактометрии. Электронно-микроскопическое исследование осуществляли при помощи растрового электронного микроскопа РЭМ100У в интервале увеличений от ×1000 до ×20000.
На рис. 2 приводятся микрофотографии с увеличением ×2000 и ×5000.На рис. 2, а, б приведены электронные микрофотографии структуры твердой фазы, находящейся в равновесии с раствором, содержащим ионы ингибитора и ионы кальция в мольном соотношении Nm = 0,01. Можно видеть, что в этом случае твердая фаза представлена исключительно кристаллическими зернами столбчатого габитуса, которые образуют сростки и друзы.
Несмотря на то, что эти зерна не всегда хорошо огранены, их кристаллическое строение не вызывает сомнения. Микрофотография структуры твердой фазы, находящейся в равновесии с раствором при Nm = 0,02, представлена на рис. 2, в, г. Характерно наличие хорошо ограненных кристаллов таблитчатого и досковидного габитуса, наряду с включениями фазы, резко отличающейся по структуре и представленной агломератами зерен округлой формы.
Сфероидальный габитус этих зерен позволяет предположить изотропию их свойств, а следовательно, аморфное строение. Аналогичную структуру имеет твердая фаза, находящаяся в равновесии с раствором при Nm = 0,04 (рис. 2, д, е). В этом случае частицы с выраженным кристаллическим строением обнаружить в твердой фазе не удалось.
Таким образом, результаты электронномикроскопического исследования структуры твердой фазы позволяет предположить, что различный характер зависимости S = f(Nm) в различных интервалах Nm обусловлен тем, что в равновесии с раствором находятся твердые фазы различной структуры или их механическая смесь.В целях проверки этой гипотезы были проведены исследования дифракции рентгеновских лучей на образцах твердой фазы, полученных в экспериментах при значениях Nm = 0,01; 0,02; 0,04.
Исследования проводились на рентгеновском дифрактометре «ДРОН6» в Coизлучении с длиной волны 1,79 ангстрем. Полученные рентгендифрактограммы представлены на рис. 3.Можно видеть, что образец твердой фазы, полученный при Nm = 0,01 (рис. 3, а), имеет ярко выраженную кристаллическую структуру кальцита, что подтверждается наличием на рентгендифрактограмме узких, однозначно индицируемых дифракционных рефлексов.
Отметим, что рефлексы хорошо разрешены, в частности, в области больших углов Вульфа-Брэгга заметно разрешение дублета (1 – 2), что указывает на отсутствие напряжений, вызванных деформациями и дефектами кристаллической решетки. Рентгендифрактограмма твердой фазы, полученной при мольном соотношении Nm = 0,02, представлена на рис. 3, б. Можно отметить наличие характерных для кальцита рефлексов, выраженных, однако, гораздо слабее, чем в предыдущем случае.
Твердая фаза, образовавшаяся в системе при Nm = 0,04 (рис. 3, в), имеет рентгеноаморфную структуру. Таким образом, результаты исследования дифракции рентгеновских лучей подтверждают вывод о различии строения твердых фаз, образующихся при различных значениях мольного соотношения.
Математическая модель
Развернутое изложение математической модели, предложенное для объяснения полученных результатов, приведено в работах [1, 7, 8]. В настоящей статье приводится краткое изложение математической модели. Рассматриваемая система включает жидкую фазу L, представляющую собой раствор состава «вода–карбонат кальция–НТФК», и две находящиеся в контакте с ней фазы S1 и S2. Фаза S1 имеет кристаллическую структуру кальцита CaCO3 [9], а фаза S2 — аморфную структуру соединения со следующим химическим составом N(CH2PO3)3Ca2,5H [2].
Сечения (т.е. относительные объемные доли) фаз L, S1 и S2 обозначим, соответственно, через L, S1 и S2. Очевидно, что L + S1 + S2 = 1. Фазовые переходы происходят между жидкой фазой и каждой из твердых фаз согласно схеме S1 L S2.Химический состав твердых фаз будем считать приблизительно постоянным. Концентрации ионов в каждой фазе связаны условиями электронейтральности:
qCO3CCO3 + qCaCCa + qinhCinh = 0, (2)
где qCO3 = –2, qCa = +2, qinh = –3 — заряды соответствующих ионов (выраженные в единицах заряда протона).Относительный объем твердых фаз в рассматриваемой системе мал по сравнению с общим объемом системы: S1 << 1 и S2 << 1, так что можно положить L ≈ 1 и dL/dt ≈ 0. Приписав фазам S1 и S2 постоянный химический состав условия материального баланса можно записать в виде:
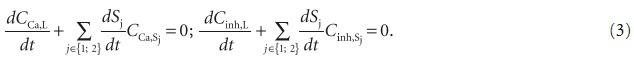
Равновесие L S1 описывается условиемCCa,LCCO3,L = PCaCO3(T).где PCaCO3(T) — произведение растворимости карбоната кальция при данной температуре T. Скорость роста или растворения кристалла, согласно экспериментальным данным [5] и теоретическим выкладкам [7, 8], может быть представлена:
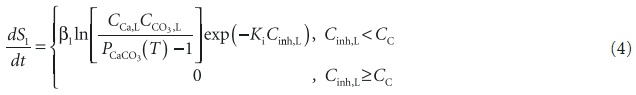
где 1 — коэффициент скорости роста кристаллической фазы, Ki — коэффициент эффективности ингибирования, учитывающий структуру и потенциальную энергию взаимодействия иона НТФК и поверхности кристалла, а также зависящий от температуры (выражение для Ki выведено в работах [7, 8], CC — критическая концентрация ионов НТФК в жидкой фазе, при которой кристаллизация полностью прекращается. Условие равновесия L S2, дается выражением (1). Для скорости роста или растворения аморфной фазы примем приближенное выражение:
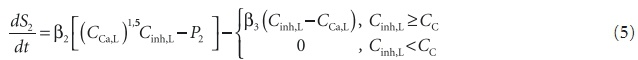
где 2, 3 — коэффициент скорости роста и растворения аморфной фазы. Последний член учитывает описанную в литературе [6] возможность образования растворимого кальциевого комплекса ингибитора при их эквимольном взаимодействии, что приводит к растворению кальцийсодержащей аморфной фазы, вследствие чего при Cinh,L > CCa,L в выражении (6) решающую роль играет последний член, и знак dS2/dt всегда неположителен.
Уравнения (2–5) образуют замкнутую систему обыкновенных дифференциальных уравнений, которая (с соответствующими начальными условиями) представляет собой математическую модель процесса конкурентного роста кристаллической и аморфной фаз в системе «вода–карбонат кальция–НТФК».Мы ограничились проведением вычислительных экспериментов для модели (2–5)с начальными условиями S1(0) = 0, S2(0) = 0, т.е. система в начальный момент времени считалась однофазной, что соответствовало условиям эксперимента.
Параметры математической модели для проведения расчетов в большинстве своем были взяты из [2, 9–12]. В качестве подгоночных параметров были оставлены кинетические коэффициенты 1, 2 и 3, которые были уточнены по критерию наилучшего согласия результатов моделирования с экспериментальными данными. Уточненные значения составили 1 ≈ 10–9,2 ≈ 10–3 (моль/дм3)–7/2, 3 ≈ 10–3 моль/дм3.Результаты моделирования (красные точки) и экспериментально полученные данные (синие точки) графически совмещены на рис. 1.
Можно отметить, что согласие результатов математического моделирования с результатами эксперимента неплохое. Далее с использованием разработанной математической модели была проведена серия вычислительных экспериментов, целью которых было выяснить влияние ингибитора на процессы фазообразования в интервале значений начальной концентрации ионов кальция [2,5; 25] моль/м3. Значения начальной концентрации НТФК выбирались таким образом, чтобы мольное соотношение Nm = Cinh/CCa пробегало интервал [0,001; 1].
Результаты моделирования представлены на рис. 4.Можно видеть, что при всех значениях начальной концентрации ионов кальция в системе зависимость суммарного сечения твердых фаз S = S1 + S2 имеет бимодальный характер, что в целом подтверждает качественные данные о характере зависимости S = f(Nm), приведенные в работе [6]. Максимум S в области малых значений Nm обусловлен образованием кристаллической фазы (синяя кривая) из-за недостаточного ингибирования кристаллизации.
Минимум S объясняется эффективным ингибированием кристаллизации в этой области значений Nm, в то же время аморфная твердая фаза термодинамически неустойчива из-за невысокой концентрации ионов кальция и НТФК в жидкой фазе. Максимум S в области более высоких значений Nm объясняется термодинамической устойчивостью аморфной фазы (красная кривая) в этой области значений концентрации ионов НТФК.
При дальнейшем повышении концентрации ионов НТФК равновесие смещается в сторону растворения аморфной твердой фазы за счет образования растворимых соединений кальция с ионами НТФК. Наибольшее значение для практики имеет область значений Nm, в которой значение S минимально (в идеале — равно нулю) или близко к минимальному, т.к. этим обеспечивается наименьшая величина солеотложений в системе.
На рис. 4, а–в, можно отметить, что при значениях начальной концентрации ионов кальция до 5 моль/м3 минимальное значение сечения твердой фазы S равно нулю, следовательно, в этих случаях возможно полное ингибирование процесса солеотложения. По мере повышения начальной концентрации ионов кальция от 1 до 5 моль/м3 диапазон значений Nm, в котором S = 0, сужается. При моль/м3интервал оптимальных значений Nm стягивается в точку.
При больших значениях концентрации ионов кальция минимальное значение сечения твердой фазы больше нуля, т.е. полное ингибирование солеотложения в этом случае невозможно (рис. 4, г).
Заключение
Приведенные в настоящей работе экспериментальные данные в целом подтверждают бимодальный характер зависимости равновесного содержания твердой фазы от дозировки ингибитора. Это связано с различным строением твердых фаз, причем при малых концентрациях ионов ингибитора в системе образуется кристаллическая фаза, а при высоких значениях концентрации ионов ингибитора — аморфная фаза.
Области существования кристаллической и аморфной фаз могут перекрываться. На основании теоретических представлений и экспериментальных данных предложена математическая модель конкурентного образования кристаллической и аморфной фаз в условиях адсорбционного ингибирования кристаллизации. Результаты вычислительных экспериментов показывают согласие с литературными и экспериментальными данными и допускают объяснение с позиций теоретических представлений о механизме и основных закономерностях действия ингибиторов кристаллизации.
Область концентрации ионов кальция, в пределах которой возможно полное ингибирование солеотложения, ограничена. При этом интервал значений концентрации НТФК, в пределах которого достигается полное ингибирование солеотложения, с увеличением концентрации ионов кальция сокращается, поэтому при более высоком содержании кальция в системе необходимо более точное дозирование ингибитора.
При больших значениях концентрации ионов кальция полное ингибирование солеотложения невозможно. Однако, изменяя концентрацию ионов НТФК, можно менять распределение ионов кальция между кристаллической и аморфной фазами.
